

Help Unlock a Child’s Superpowers!
Make a gift and a kid like Naomi will get the book "What Makes a Superhero?"
Give Today
Pediatric Specialty Care in Greenville

We are honored to be selected for the Guardian of Excellence for Patient Experience and Physician Engagement awards by Press Ganey.
About Shriners Children's Greenville
Home to the largest team of pediatric orthopedic surgeons in the Carolinas, Shriners Children’s Greenville has been recognized by community vote as “Best in the Upstate” for Best Hospital, Best Pediatric Clinic, Best Orthopedics Center and Best Place to Work.
Specialty Care Provided at Greenville


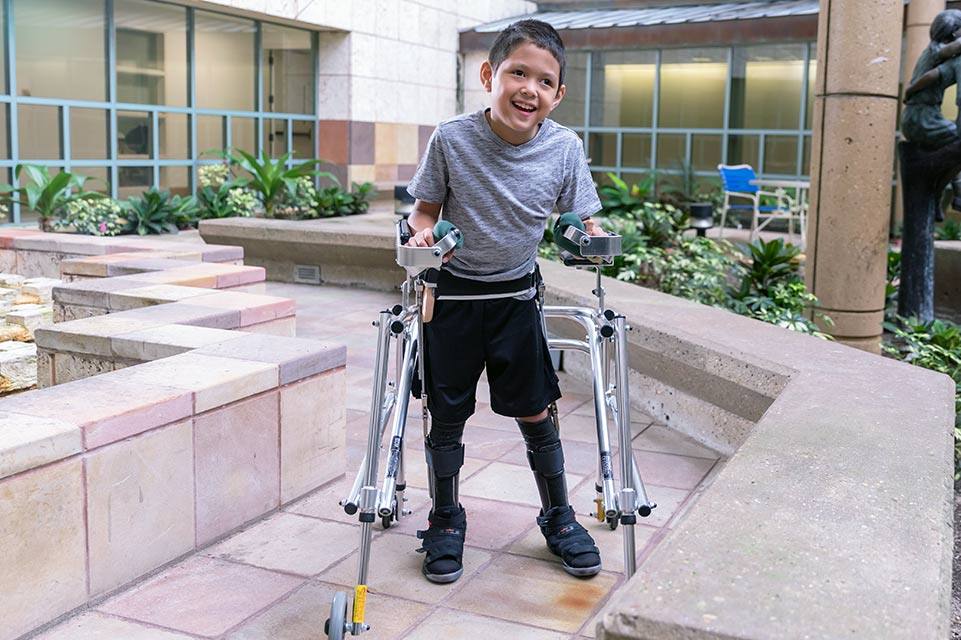



We Understand the Unique Medical Needs of Children
We provide vital, pioneering treatment from birth to age 18. Here, children have the opportunity to be evaluated and treated by doctors recognized as the best by their peers.
What's Happening at Shriners Children's Greenville




Help Us Provide the Gift of Hope and Healing
Every gift has an impact, which is why there are multiple ways you can help.
Keep In Touch
Join our mailing list to stay up to date on everything that's happening at Shriners Children's.